| Dr Kevin Sheridan | Postdoctoral Fellow, UCD CARPostdoctoral Fellow, UCD CAR |
|---|
This project is funded by the National Children's Research Centre
Auto-inflammatory conditions commonly cause patients to have episodes of unprovoked inflammation that can affect one or multiple organs in the body. These conditions can be caused by a mutation (change) in a gene (part of their DNA) that controls what is called the innate immune system. The innate immune system manages the body’s rapid response to threats; it acts as the first responder to an infection caused by pathogens; pathogens are germs that can cause disease, such as viruses and bacteria. In response to a pathogen, inflammatory signals are released in the infected area which prepares the body to fight the infection. In patients with an auto-inflammatory condition, the body acts as if an infection were present, even when there is none. Although many of these auto-inflammatory conditions are very severe, they are often very responsive to new anti-inflammatory medicines. These medicines work by blocking the chemical signals that trigger inflammation in our bodies. Identifying the genetic cause (i.e. mutations in patient DNA) of these conditions can help doctors choose the best treatment for patients.
Behçet’s Disease (BD) is an auto-inflammatory condition where patients’ symptoms include ulcers in the mouth or genitals, inflammation in the eye, rashes and, less commonly, inflammation of the bowels, large blood vessels and the brain. It is believed to be caused by a combination of a large number of variations in the DNA sequence, each contributing small but accumulative biological effects, and unknown environmental exposure(s).
A BD-like illness, affecting both adults and children, has recently been identified in six families by researchers in the US. It is caused by changes in one of the genes that controls inflammation, called TNFAIP3. We have identified five Irish families with a similar BD-like illness. One of these families, a large three generation family with four affected individuals, was first selected for analysis of their DNA. Over 19,000 genes in their DNA were analysed and although they have normal TNFAIP3, a mutation in a gene called RELA was found in all affected family members. This gene is considered a master regulator of inflammation. The mutation prevents RELA from functioning properly and as a result, patients experience frequent episodes of inflammation and ulcers.
In the next part of the study we will perform DNA analysis to find the mutations causing disease in the other four families. We will also perform experiments in the laboratory, some of which will study the blood cells of affected and unaffected family members, to understand how the identified mutations can disrupt the normal processes that the genes are involved in.
Importantly, our study will focus on uncovering evidence that allows doctors to choose the most appropriate treatment for each patient (targeted treatment). This will lead to earlier and better disease control, therefore preventing bodily damage and improving the quality of life for the affected patients.
Behçet’s disease (BD) is an auto-inflammatory condition involving recurrent episodes of oral and genital ulceration, inflammation in the eye, rashes, and less frequent involvement of the bowels, large blood vessels and brain. It is believed to be caused by a combination of a large number of genetic variants, each contributing small but accumulative biological effects, and unknown environmental exposure(s). A BD-like illness, affecting both adults and children, has recently been identified in several families by researchers in the US and shown to be related to mutations in two NF-κB pathway genes.
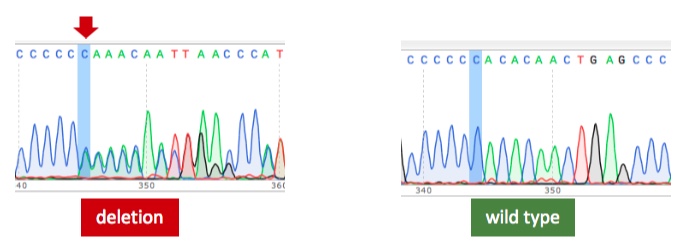
We have identified 8 Irish families with a BD-like illness and multiple ‘sporadic’ cases of a BD-like illness with atypical features, primarily involving childhood-onset chronic oral and genital ulcers. In the largest, a three generation family, whole exome sequencing revealed segregation of a mutation in RELA (p65) with the condition. The mutation involves a cytosine deletion causing a frameshift resulting in a truncated protein of 492 amino acids, compared with wild-type RELA of 551 amino acids. Crucially, the mutation interrupts the two C-terminal RELA transactivating domains. The truncated RELA protein is expressed at similar levels as the wild-type in PBMCs.
Our hypothesis is that mutations in NF-κB pathway genes are the basis of the mucocutaneous syndrome in the Irish families. Our first objective will be to determine the biological effects of the RELA mutation on NF-κB pathway activities such as inflammation and apoptosis. Secondly, whole exome sequencing of the four other families will be performed to identify potentially causative mutations. The final objective will be to undertake appropriate biological studies to determine genotype-phenotype associations.
An important outcome will be to find evidence that allows paediatrians to choose the most appropriate treatment for each child and family. This will lead to earlier and better disease suppression, preventing tissue damage and improving the quality of life of affected children.
This work will be completed by Drs Kevin Sheridan & Daire O’Leary in collaboration with Dr Eoin Cummins, Prof Gerry Wilson, Prof Cormac Taylor at UCD Conway Institute and Dr Orla Killeen at Our Lady's Children's Hospital Crumlin. Collaborating on this project is Dr Alexander Fraser, Consultant Rheumatologist at University Hospital Limerick.