
Investigating the interplay of Epigenetics and Cancer Signalling in acquired Drug Resistance in Melanoma
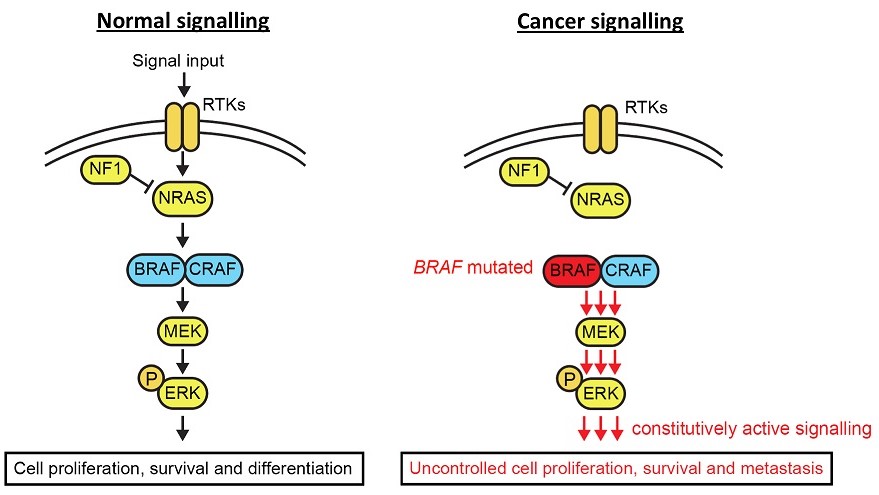
Figure 1. The MAPK signalling pathway in healthy and cancerous cells
My research has been centred around the development of novel approaches for studying protein and gene regulatory networks, with a particular focus on proteins involved in the epigenetic regulation of gene expression. In Systems Biology Ireland (SBI) I am using this expertise in methodology development and epigenetics to tackle the issue of how melanoma cells are able to rapidly develop resistance to current targeted therapies.
Metastatic melanoma is the term for late stage melanoma (or skin cancer), where the cancerous cells have spread from their original site and into other organs. It is one of the most aggressive forms of cancer and, worryingly, a cancer whose incidence is on the rise. Melanoma is primarily driven by mutations in the mitogen-activated protein kinase (MAPK) signalling pathway. The MAPK pathway is composed of a number of proteins which, in response to signalling from surrounding cells or the cell itself, activate one another in a cascading manner ultimately leading to the activation of genes responsible for driving processes such as cell growth and survival1 (Figure 1). In healthy tissues, this pathway is tightly regulated so that cells only grow in response to the correct signals, and when that stimulus is removed, stop growing. However, when the proteins in this pathway get altered in cancer, this regulation gets disrupted and results in the cancer cells being able to grow indefinitely and without the correct signals. For instance, in melanoma a small mutation in the gene that encodes the protein BRAF results in this protein being able to remain active in the absence of stimulation, and continuously send signals to the cancer cells to keep growing and multiplying.
The MAPK signalling pathway is one of the most frequently mutated pathways, altered in about 30% of all cancer types. In melanoma alone, 50% of patients carry a mutation in the BRAF gene, while another 30% carry mutations in the NRAS gene2. Due to this, a large focus of melanoma research has historically been on developing drugs to inhibit this pathway, and thereby stop the growth and survival of cancer cells. Drugs targeting the mutant form of BRAF were widely successful, where patients saw significant tumour shrinkage and clearance within as little as 2 weeks. However, melanoma remains a deadly disease with extremely poor prognosis. Despite initial success in treating patients, the development of resistance to these drugs occurs invariably and within as little as 6 months of treatment. This resistance can occur through numerous different mechanisms, but generally involves the rewiring of signalling networks in order to bypass the drugs’ inhibition of MAPK signalling, essentially giving cancer cells a detour around the drug (Figure 2). This can involve either secondary mutations in the MAPK pathway itself, or can involve the activation of alternative signalling pathways which can compensate for the lack of signalling through the MAPK pathway and continue to drive cancer cell growth and survival3.
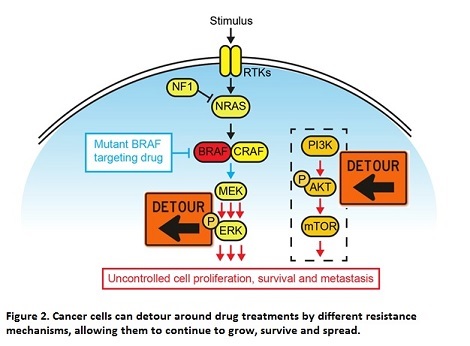
The mechanisms involved in the activation of alternate pathways is a key area of research for the development of new therapies to treat melanoma.
My research aims to elucidate whether epigenetic genes and their proteins are interacting with cancer signalling pathways and facilitating the development of drug resistance in melanoma.
So what exactly is epigenetics? Well to understand this we have to look at DNA, the genes within it, and how this is housed in our cells. Every single cell in our bodies, whether it’s a simple skin cell or a complicated brain cell, contain exactly the same DNA. What makes these cells different is how the genes in that DNA are expressed: which are turned on and which are turned off at any given moment. Genes are the parts of our DNA that code for proteins, and proteins are basically the workhorses of our cells carrying out all the reactions necessary for our cells to function. So, it is the unique repertoire of different combinations of actively expressed genes, and their encoded proteins, that essentially make up each different type of cell.
But what dictates which genes to turn off and which to turn on, and how is achieved? What you might not realise is that any single cell in our body contains so much DNA (nearly 2 m if you could stretch it out!) that we have had to develop very intricate ways of packing all that material into our microscopic cells. To do this, our DNA is wrapped around proteins called histones and this forms a dense fibre-like structure called chromatin. This chromatin also then gets even more densely wrapped around itself, and eventually after all that compaction we get these structures called chromosomes. This compaction of DNA into chromatin allows us to fit all that DNA into a single cell, but it also creates an important way in which our cells can control the expression of genes, by changing the accessibility to these genes (Figure 3).
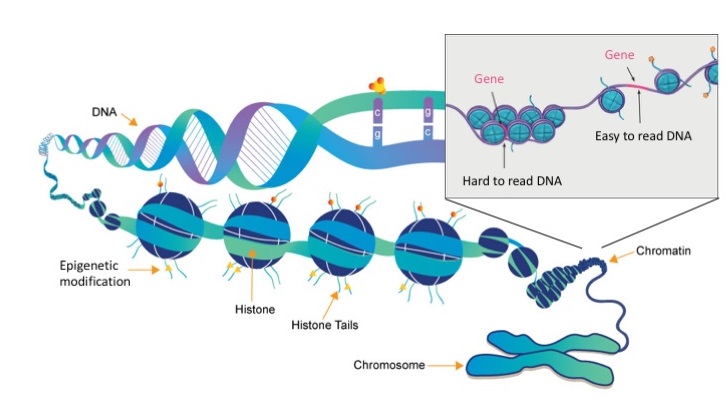
Figure 3. Organisation of DNA and histone proteins into chromosomes (images modified from www.whatisepigenetics.com and the National Institutes of Health)
Throughout our lives, and even just the life of a single cell, the genes contained in our DNA need to be carefully controlled – sometimes silencing them to prevent them from being expressed, and at other times activating them so that they can express their proteins and carry out their functions. Our cells have developed very sophisticated mechanisms to do this, and at the core of this is a process we call epigenetic regulation of gene expression.
What this involves, put simply, is a collection of different proteins which act together to bind to chromatin and manipulate the degree to which it is compacted. If chromatin is very densely packed, access to these genes is restricted and so the proteins which activate their expression cannot bind, and therefore these genes are effectively silenced. Conversely, if the chromatin compaction is relaxed, opening it up, proteins can now gain access to the genes and turn them on.
The proteins involved in manipulating chromatin compaction are called epigenetic proteins, and they can alter chromatin in a number of different ways4. Some epigenetic proteins generate little ‘tags’ on the histone proteins which (together with the DNA) form chromatin, and these tags can then either directly influence the way in which the chromatin gets compacted or can serve as binding sites for other epigenetic proteins, such as those which can physically manipulate chromatin structure through a process called ‘chromatin remodelling’. (Figure 4). An important feature of epigenetic regulation is that all these modifications are reversible – so that the resulting silencing or activation of genes is not permanent and can be changed as required by the cell. For instance, when stem cells develop into specific types of cells such as brain cells, genes involved in neuronal development need to be activated, while genes that previously made them stem cells have to be silenced. It is through epigenetic regulation like this that cells, despite being identical on the DNA level, can become vastly different and specialised in form and function.
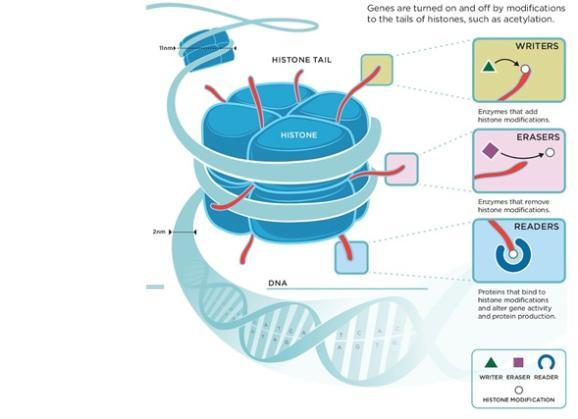
Figure 4. Epigenetic modification of histone protein tails (image taken from www.zenithepigenetics.com, copyright of Richard E. Ballermann)
The alteration of genes involved in epigenetics are increasingly becoming a focus in cancer research, as recent work has shown that epigenetic genes are frequently mutated in cancer4 and can contribute to the development of drug resistance in tumours. Because of this, and because of the previously mentioned reversibility of their regulation mechanisms, epigenetic genes and the proteins they encode offer promising candidates for new therapeutic targets in the treatment of cancer and combating tumours’ resistance to current drugs.
My work in Systems Biology Ireland is focused on investigating whether epigenetics plays a role in the development of drug resistance in metastatic melanoma. To do this, I am applying techniques developed during my PhD, which were recently published in Molecular Cell5, where we combined proteomic and genomic approaches to study how protein-protein interactions can influence the activity of a group of epigenetic regulators called the ‘Polycomb group’ proteins. These proteins are fundamentally important to the development of cells, and have also been implicated in driving a number of cancers and developmental disorders. By understanding how their activity is regulated by other proteins we have provided new insights into the mechanisms underlying these processes and diseases.
I now aim to use approaches similar to those in this publication to study the role of epigenetics in the development of drug resistance in metastatic melanoma. By using a combination of published literature and mutant BRAF protein interaction data, I am selecting a panel of epigenetic genes to target in melanoma cell lines, both in the presence and absence of the mutant BRAF inhibitor Vemurafenib. By perturbing the expression of these genes, I will determine whether any of these epigenetic genes can impact the development of Vemurafenib drug resistance and ascertain whether this effect is due to increased signalling activity. Epigenetic-targeting drugs are already both in development and clinically approved for use. Therefore, the hope is that we can provide new rationale for combining existing epigenetic inhibitors with the gold standard Raf inhibitors, to provide better prospects for melanoma patients.
References
- Zhang W & Liu HT. 2002. “MAPK signal pathways in the regulation of cell proliferation in mammalian cells”. Cell Research 12;9-18.
- Burotto M, Chiou VL, Lee JM, Kohn EC. 2014. “The MAPK pathway across different malignancies: a new perspective”. Cancer Nov 15;120(22):3446-56
- Arozarena I & Welklbrock C. 2017. “Overcoming resistance to BRAF inhibitors”. Ann. Transl. Med. Oct;5(19):387
- Dawson, MA & Kouzarides T. 2012. “Cancer Epigenetics: From Mechanism to Therapy”. Cell. 2012 Jul 6;150(1):12-27.
- Streubel G, Watson A, Jammula SG, Scelfo A, Fitzpatrick DJ, Oliviero G, McCole R, Conway E, Glancy E, Negri GL, Dillon E, Wynne K, Pasini D, Krogan NJ, Bracken AP, Cagney G. 2018. Mol. Cell. pr 19;70(2):371-379.e5.
About the author

| Dr. Ariane Watson was awarded a postdoctoral fellowship in 2017 under the Marie Skłodowska-Curie COFUND Action through System Biology Ireland's TOPMed10 programme. Under the supervision of Prof. Walter Kolch and Dr. Colm Ryan, she is currently investigating the role of crosstalk between epigenetic machinery and canonical signalling pathways in facilitating the development of drug resistance in late stage melanoma. |

