
Modeling Treatments for RAS- and RAF-mutant Cancers
Cancers, particularly melanomas, bearing oncogenic NRAS and HRAS mutations are currently considered undruggable. Efforts to target oncogenic NRAS and HRAS directly or inhibit RAS downstream pathways, including RAF kinases, have limited clinical success. The lack of response of RAS-mutant melanomas to clinically used RAF inhibitors is related to the increase in homo- and heterodimerization of RAF kinases, which is driven by oncogenic RAS and also leads to the paradoxical activation of ERK signaling by RAF inhibitors. For melanomas carrying wild type RAS and mutant BRAFV600E, RAF inhibitors, such as Dabrafenib and Vemurafenib, often given in combination with MEK inhibitors (Trametinib), result in a good initial response. Unfortunately, resistance inevitably develops, in most cases driven by the increase in RAF homo- and hetero-dimerization or by acquisition of oncogenic RAS mutations (Durrant and Morrison, 2017).
Drug resistance and thermodynamic principles
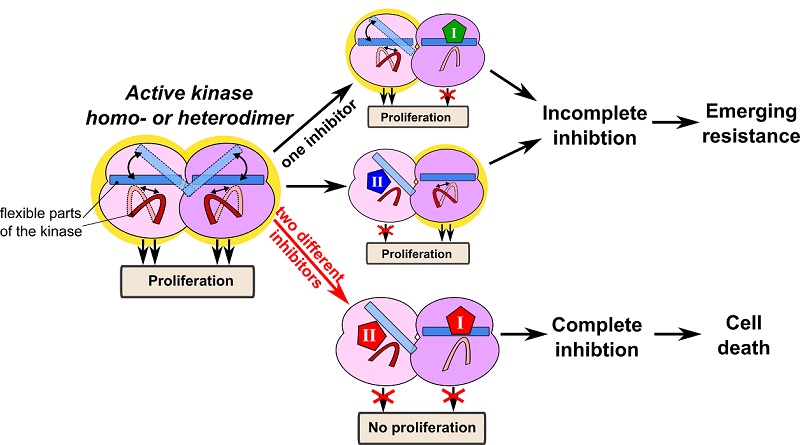
Interestingly, the first insight into how to overcome the drug resistance caused by kinase dimerization has come from mathematical modeling. It appears that the resistance of RAF dimers to drugs results from fundamental thermodynamic principles (Kholodenko, 2015). Mutant RAS and current clinically used RAF inhibitors induce RAF dimers, in which the protomers acquire different conformations (Jambrina et al, 2016). In this constellation one protomer is drug bound and allosterically activates the other drug-free protomer conferring resistance. The emergence of different drug affinities between monomers and protomers in a dimer has been enigmatic, but has been explained by thermodynamics. Overcoming enhanced RAF dimerization, especially in RAS-mutant cancers, and the resulting resistance is a challenge for drug design. For instance, paradox breaker RAF inhibitors that do not increase RAF dimerization have been developed, yet they do not inhibit RAF dimers (Zhang, C. et al, 2015). All other RAF inhibitors, which are able to inhibit RAF dimers, induce paradoxical ERK activation at some doses. Exploiting thermodynamic principles, we showed how two RAF inhibitors, ineffective on their own, can inhibit RAF homo-or heterodimers when combined at lower doses than either inhibitor applied alone, thus abolishing drug resistance (Kholodenko, 2015). A key question is how to find best inhibitor combinations to overcome oncogenic RAS-driven resistance.
Can mechanistic mathematical models help us?
To enable mechanistic models to predict ways to effectively inhibit oncogenic RAS-driven ERK signaling, while disabling or delaying signal recovery, growth and drug resistance, these models have to account for both: 1) Conformational selectivity of kinase inhibitors – affecting structural, thermodynamic and kinetic properties of RAF/MEK/ERK kinases; and 2) Emergent network properties determined by cell-specific protein levels, mutations, and regulatory loops. Our team at the institute of Systems Biology Ireland (University College Dublin, Ireland) together with international collaborators in the UK, US, and Greece, used a combined experimental and computational approach to overcome resistance to RAF inhibitors in RAS-mutant cancers. We developed a next-generation computer model that not only integrates the protein-protein interaction and network architecture, but also includes the thermodynamics and kinetics of drug- protein interactions, spontaneous intra-molecular motions of proteins, structural conformations of the protein targets and inhibitors, and post-translational modifications of the signaling molecules (Rukhlenko et al, 2018). By taking into account conformational preferences of inhibitors (stabilization of active, inactive, or distorted kinase conformations) and their thermodynamic and kinetic properties, this model demonstrated a number of unexpected features of network responses to different types of RAF inhibitors. For individual protein-abundance and mutational profiles, the model predicted and experiments corroborated synergistic combinations of conformation-selective RAF inhibitors, which effectively suppressed not only oncogenic RAF, but also oncogenic RAS signaling, cancer cell proliferation and focus formation. Importantly, we demonstrated which specific RAF inhibitor combinations have to be applied to overcome oncogenic RAS signaling. Model predictions go beyond malignant melanomas to all cancers bearing RAS-mutations (e.g. colorectal cancer, pancreatic cancer, non-small cell lung cancer, etc.).
What’s next?
The same thermodynamic principle of targeting kinase dimers by two inhibitors that may target the same enzyme pocket but with distinct conformation selectivity is applicable to multiple kinases whose activation cycle includes dimerization or oligomerization, such as ERBB and JAK kinases. Our computational models and experiments show synergy between different conformation-selective ERBB inhibitors in HER2-positive breast cancer cells and different conformation-selective JAK inhibitors in IL7R-mutant T-cell acute lymphatic leukemia cells. We suggest that targeting an enzyme with two structurally different small molecule inhibitors that bind to the same pocket in the target can be a new widely applicable strategy to overcome resistance induced by enzyme dimerization or oligomerization. We also present an approach how to develop mathematical models to analyze best drug combinations specifically for each case. The theoretical and experimental data also suggest novel potential treatment options for RASopathies, which are developmental disorders caused by germline mutations in the components of the RAS/RAF/MEK/ERK pathway. Synergistic combinations of small doses of RAF inhibitors could be applied to RASopathies with germline mutations in HRAS, NRAS, KRAS, SOS1, SHOC2, RASA, NF1 and RAF mutations. Modeling can predict the inhibitor doses that will modulate the ERK pathway activity restoring it to the norm, while avoiding toxicity. Finally, populating next-generation mechanistic models with specific cellular protein abundance and mutational profiles, information about conformation selective inhibitors and mechanisms of emerging resistance to single drugs will allow researchers to target different RAS-mutant cancers, such as pancreatic cancer. We are looking for academia and/or industry partners who would be interested in this approach to find best drug combinations and could test our model predictions in vivo, using cell line-derived xenografts, patient-derived xenografts, and animal models.
References
- Durrant, D. E. & Morrison, D. K. (2018) Targeting the Raf kinases in human cancer: the Raf dimer dilemma. British journal of cancer 118, 3-8, doi:10.1038/bjc.2017.399.
- Kholodenko, B.N. (2015) Drug Resistance Resulting from Kinase Dimerization Is Rationalized by Thermodynamic Factors Describing Allosteric Inhibitor Effects. Cell reports 12, 1939-1949, doi:10.1016/j.celrep.2015.08.014.
- Jambrina et al. (2016) Phosphorylation of Raf Kinase Dimers Drives Conformational Changes that Facilitate Transactivation. Angewandte Chemie 55, 983-986, doi:10.1002/anie.201509272.
- Rukhlenko et al. (2018) Dissecting Raf Inhibitor Resistance by Structure-based Modeling Reveals Ways to Overcome Oncogenic Ras Signaling, Cell Syst. 7, 161-179 e14.
- Zhang, C. et al. (2015) Raf inhibitors that evade paradoxical MAPK pathway activation. Nature 526, 583-586, doi:10.1038/nature14982.

About the Author
Trained as a biophysicist, Boris Kholodenko did postdoctoral training with A. Zhabotinsky (known for the oscillatory Belousov-Zhabotinsky reaction). His career has focused on mathematical and experimental analysis of biological networks. Together with Walter Kolch he now co-directs Systems Biology Ireland (SBI) at University College Dublin.

